Identify TCR/BCR Clones in a Neighborhood Around Each Associated Sequence
findAssociatedClones.RdPart of the workflow
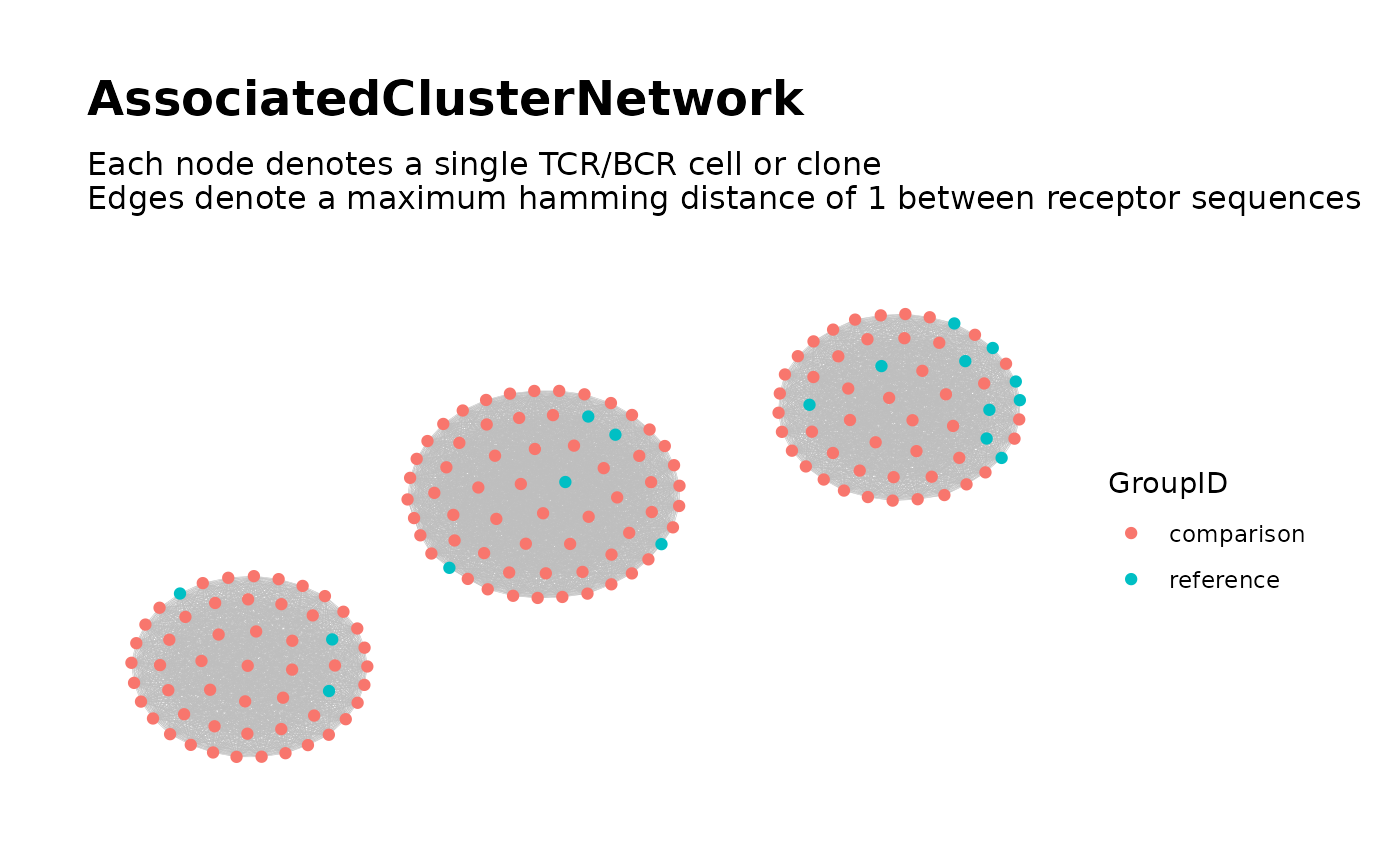
Searching for Associated TCR/BCR Clusters.
Intended for use following findAssociatedSeqs()
and prior to
buildAssociatedClusterNetwork().
Given multiple samples of bulk Adaptive Immune Receptor Repertoire Sequencing (AIRR-Seq) data and a vector of associated sequences, identifies for each associated sequence a global "neighborhood" comprised of clones with TCR/BCR sequences similar to the associated sequence.
Usage
findAssociatedClones(
## Input ##
file_list, input_type,
data_symbols = NULL,
header, sep, read.args,
sample_ids =
paste0("Sample", 1:length(file_list)),
subject_ids = NULL,
group_ids,
seq_col,
assoc_seqs,
## Neighborhood Criteria ##
nbd_radius = 1,
dist_type = "hamming",
min_seq_length = 6,
drop_matches = NULL,
## Output ##
subset_cols = NULL,
output_dir,
output_type = "rds",
verbose = FALSE
)Arguments
- file_list
A character vector of file paths, or a list containing
connectionsand file paths. Each element corresponds to a single file containing the data for a single sample. Passed toloadDataFromFileList().- input_type
A character string specifying the file format of the sample data files. Options are
"table","txt","tsv","csv","rds"and"rda". Passed toloadDataFromFileList().- data_symbols
Used when
input_type = "rda". Specifies the name of each sample's data frame within its respective Rdata file. Passed toloadDataFromFileList().- header
For values of
input_typeother than"rds"and"rda", this argument can be used to specify a non-default value of theheaderargument toread.table(),read.csv(), etc.- sep
For values of
input_typeother than"rds"and"rda", this argument can be used to specify a non-default value of thesepargument toread.table(),read.csv(), etc.- read.args
For values of
input_typeother than"rds"and"rda", this argument can be used to specify non-default values of optional arguments toread.table(),read.csv(), etc. Accepts a named list of argument values. Values ofheaderandsepin this list take precedence over values specified via theheaderandseparguments.- sample_ids
A character or numeric vector of sample IDs, whose length matches that of
file_list. Each entry is assigned as the sample ID to the corresponding entry offile_list.- subject_ids
An optional character or numeric vector of subject IDs, whose length matches that of
file_list. Used to assign a subject ID to each sample.- group_ids
A character or numeric vector of group IDs whose length matches that of
file_list. Used to assign each sample to a group. The two groups represent the levels of the binary variable of interest.- seq_col
Specifies the column of each sample's data frame containing the TCR/BCR sequences. Accepts a character string containing the column name or a numeric scalar containing the column index.
- assoc_seqs
A character vector containing the TCR/BCR sequences associated with the binary variable of interest.
- nbd_radius
The maximum distance (based on
dist_type) between an associated sequence and other TCR/BCR sequences belonging to its neighborhood. Lower values require sequences to be more similar to an associated sequence in order to belong to its neighborhood.- dist_type
Specifies the function used to quantify the similarity between sequences. The similarity between two sequences determines their pairwise distance, with greater similarity corresponding to shorter distance. Valid options are
"hamming"(the default), which useshamDistBounded(), and"levenshtein", which useslevDistBounded().- min_seq_length
Clones with TCR/BCR sequences below this length will be removed. Passed to
filterInputData()when loading each sample.- drop_matches
Passed to
filterInputData(). Accepts a character string containing a regular expression (seeregex). Checks TCR/BCR sequences for a pattern match usinggrep(). Those returning a match are dropped. By default, sequences containing any of the characters*,|or_are dropped.- subset_cols
Controls which columns of the AIRR-Seq data from each sample are included in the output. Accepts a character vector of column names or a numeric vector of column indices. The default
NULLincludes all columns. Passed tofilterInputData().- output_dir
A file path to a directory for saving the output. A valid output directory is required, since no output is returned in R. The specified directory will be created if it does not already exist.
- output_type
A character string specifying the file format to use for saving the output. Valid options are
"rda","csv","csv2","tsv"and"table". For"rda", data frames are nameddatain the R environment. For the remaining options,write.table()is called withrow.names = TRUE.- verbose
Logical. If
TRUE, generates messages about the tasks performed and their progress, as well as relevant properties of intermediate outputs. Messages are sent tostderr().
Details
For each associated sequence, its neighborhood is defined to include all
clones with TCR/BCR sequences that are sufficiently similar to the associated
sequence. The arguments dist_type and nbd_radius control how the
similarity is measured and the degree of similarity required for neighborhood
membership.
For each associated sequence, a data frame is saved to an individual file. The data frame contains one row for each clone in the associated sequence's neighborhood (from all samples). It includes variables for sample ID, group ID and (if provided) subject ID, as well as variables from the AIRR-Seq data.
The files saved by this function are intended for use with
buildAssociatedClusterNetwork(). See the
Searching for Associated TCR/BCR Clusters
article on the package website for more details.
References
Hai Yang, Jason Cham, Brian Neal, Zenghua Fan, Tao He and Li Zhang. (2023). NAIR: Network Analysis of Immune Repertoire. Frontiers in Immunology, vol. 14. doi: 10.3389/fimmu.2023.1181825
Searching for Associated TCR/BCR Clusters article on package website
Author
Brian Neal (Brian.Neal@ucsf.edu)
Examples
set.seed(42)
## Simulate 30 samples from two groups (treatment/control) ##
n_control <- n_treatment <- 15
n_samples <- n_control + n_treatment
sample_size <- 30 # (seqs per sample)
base_seqs <- # first five are associated with treatment
c("CASSGAYEQYF", "CSVDLGKGNNEQFF", "CASSIEGQLSTDTQYF",
"CASSEEGQLSTDTQYF", "CASSPEGQLSTDTQYF",
"RASSLAGNTEAFF", "CASSHRGTDTQYF", "CASDAGVFQPQHF")
# Relative generation probabilities by control/treatment group
pgen_c <- matrix(rep(c(rep(1, 5), rep(30, 3)), times = n_control),
nrow = n_control, byrow = TRUE)
pgen_t <- matrix(rep(c(1, 1, rep(1/3, 3), rep(2, 3)), times = n_treatment),
nrow = n_treatment, byrow = TRUE)
pgen <- rbind(pgen_c, pgen_t)
simulateToyData(
samples = n_samples,
sample_size = sample_size,
prefix_length = 1,
prefix_chars = c("", ""),
prefix_probs = cbind(rep(1, n_samples), rep(0, n_samples)),
affixes = base_seqs,
affix_probs = pgen,
num_edits = 0,
output_dir = tempdir(),
no_return = TRUE
)
#> [1] TRUE
## Step 1: Find Associated Sequences ##
sample_files <-
file.path(tempdir(),
paste0("Sample", 1:n_samples, ".rds")
)
group_labels <- c(rep("reference", n_control),
rep("comparison", n_treatment))
associated_seqs <-
findAssociatedSeqs(
file_list = sample_files,
input_type = "rds",
group_ids = group_labels,
seq_col = "CloneSeq",
min_seq_length = NULL,
drop_matches = NULL,
min_sample_membership = 0,
pval_cutoff = 0.1
)
head(associated_seqs[, 1:5])
#> ReceptorSeq fisher_pvalue shared_by_n_samples samples_g0 samples_g1
#> 8 CSVDLGKGNNEQFF 1.052106e-05 18 3 15
#> 7 CASSGAYEQYF 1.157316e-04 17 3 14
#> 4 CASSEEGQLSTDTQYF 5.197401e-03 10 1 9
#> 5 CASSIEGQLSTDTQYF 6.559548e-02 16 5 11
## Step 2: Find Associated Clones ##
dir_step2 <- tempfile()
findAssociatedClones(
file_list = sample_files,
input_type = "rds",
group_ids = group_labels,
seq_col = "CloneSeq",
assoc_seqs = associated_seqs$ReceptorSeq,
min_seq_length = NULL,
drop_matches = NULL,
output_dir = dir_step2
)
## Step 3: Global Network of Associated Clusters ##
associated_clusters <-
buildAssociatedClusterNetwork(
file_list = list.files(dir_step2,
full.names = TRUE
),
seq_col = "CloneSeq",
size_nodes_by = 1.5,
print_plots = TRUE
)