Identify TCR/BCR Sequences Associated With a Binary Variable
findAssociatedSeqs.RdPart of the workflow Searching for Associated TCR/BCR Clusters.
Given multiple samples of bulk Adaptive Immune Receptor Repertoire Sequencing (AIRR-Seq) data and a binary variable of interest such as a disease condition, treatment or clinical outcome, identify receptor sequences that exhibit a statistically significant difference in frequency between the two levels of the binary variable.
findAssociatedSeqs() is designed for use when each sample is stored in a
separate file. findAssociatedSeqs2() is designed for use with a single data
frame containing all samples.
Usage
findAssociatedSeqs(
## Input ##
file_list,
input_type,
data_symbols = NULL,
header, sep, read.args,
sample_ids = deprecated(),
subject_ids = NULL,
group_ids,
groups = deprecated(),
seq_col,
freq_col = NULL,
## Search Criteria ##
min_seq_length = 7,
drop_matches = "[*|_]",
min_sample_membership = 5,
pval_cutoff = 0.05,
## Output ##
outfile = NULL,
verbose = FALSE
)
findAssociatedSeqs2(
## Input ##
data,
seq_col,
sample_col,
subject_col = sample_col,
group_col,
groups = deprecated(),
freq_col = NULL,
## Search Criteria ##
min_seq_length = 7,
drop_matches = "[*|_]",
min_sample_membership = 5,
pval_cutoff = 0.05,
## Ouptut ##
outfile = NULL,
verbose = FALSE
)Arguments
- file_list
A character vector of file paths, or a list containing
connectionsand file paths. Each element corresponds to a single file containing the data for a single sample. Passed toloadDataFromFileList().- input_type
A character string specifying the file format of the sample data files. Options are
"table","txt","tsv","csv","rds"and"rda". Passed toloadDataFromFileList().- data_symbols
Used when
input_type = "rda". Specifies the name of each sample's data frame within its respective Rdata file. Passed toloadDataFromFileList().- header
For values of
input_typeother than"rds"and"rda", this argument can be used to specify a non-default value of theheaderargument toread.table(),read.csv(), etc.- sep
For values of
input_typeother than"rds"and"rda", this argument can be used to specify a non-default value of thesepargument toread.table(),read.csv(), etc.- read.args
For values of
input_typeother than"rds"and"rda", this argument can be used to specify non-default values of optional arguments toread.table(),read.csv(), etc. Accepts a named list of argument values. Values ofheaderandsepin this list take precedence over values specified via theheaderandseparguments.- sample_ids
![[Deprecated]](figures/lifecycle-deprecated.svg) Does nothing.
Does nothing.- subject_ids
A character or numeric vector of subject IDs, whose length matches that of
file_list. Only relevant when the binary variable of interest is subject-specific and multiple samples belong to the same subject.- group_ids
A character or numeric vector of group IDs containing exactly two unique values and with length matching that of
file_list. The two groups correspond to the two values of the binary variable of interest.- groups
-
Does nothing.
- seq_col
Specifies the column of each sample's data frame containing the TCR/BCR sequences. Accepts a character string containing the column name or a numeric scalar containing the column index.
- freq_col
Optional. Specifies the column of each sample's data frame containing the clone frequency (i.e., clone count divided by the sum of the clone counts across all clones in the sample). Accepts a character string containing the column name or a numeric scalar containing the column index. If this argument is specified, the maximum clone frequency (across all samples) for each associated sequence will be included in the content of the
labelvariable of the returned data frame.- min_seq_length
Controls the minimum TCR/BCR sequence length considered when searching for associated sequences. Passed to
filterInputData().- drop_matches
Passed to
filterInputData(). Accepts a character string containing a regular expression (seeregex). Checks TCR/BCR sequences for a pattern match usinggrep(). Those returning a match are excluded from consideration as associated sequences. It is recommended to filter out sequences containing special characters that are invalid for use in file names. By default, sequences containing any of the characters*,|or_are dropped.- min_sample_membership
Controls the minimum number of samples in which a TCR/BCR sequence must be present in order to be considered when searching for associated sequences. Setting this value to
NULLbypasses the check.- pval_cutoff
Controls the P-value cutoff below which an association is detected by Fisher's exact test (see details).
- outfile
A file path for saving the output (using
write.csv()).- verbose
Logical. If
TRUE, generates messages about the tasks performed and their progress, as well as relevant properties of intermediate outputs. Messages are sent tostderr().- data
A data frame containing the combined AIRR-seq data for all samples.
- sample_col
The column of
datacontaining the sample IDs. Accepts a character string containing the column name or a numeric scalar containing the column index.- subject_col
Optional. The column of
datacontaining the subject IDs. Accepts a character string containing the column name or a numeric scalar containing the column index. Only relevant when the binary variable of interest is subject-specific and multiple samples belong to the same subject.- group_col
The column of
datacontaining the group IDs. Accepts a character string containing the column name or a numeric scalar containing the column index. The groups correspond to the two values of the binary variable of interest. Thus there should be exactly two unique values in this column.
Details
The TCR/BCR sequences from all samples are first filtered according to minimum
sequence length and sequence content based on the specified values in
min_seq_length and drop_matches, respectively. The sequences
are further filtered based on sample membership, removing sequences appearing
in fewer than min_sample_membership samples.
For each remaining TCR/BCR sequence, a P-value is computed for Fisher's exact test of independence between the binary variable of interest and the presence of the sequence within a repertoire. The samples/subjects are divided into two groups based on the levels of the binary variable. If subject IDs are provided, then the test is based on the number of subjects in each group for whom the sequence appears in one of their samples. Without subject IDs, the test is based on the number of samples possessing the sequence in each group.
Fisher's exact test is performed using
fisher.test(). TCR/BCR
sequences with a \(P\)-value below pval_cutoff are sorted by \(P\)-value
and returned along with some additional information.
The returned ouput is intended for use with the
findAssociatedClones()
function. See the
Searching for Associated TCR/BCR Clusters
article on the package website.
Value
A data frame containing the TCR/BCR sequences found to be associated with the binary variable using Fisher's exact test (see details). Each row corresponds to a unique TCR/BCR sequence and includes the following variables:
- ReceptorSeq
The unique receptor sequence.
- fisher_pvalue
The P-value on Fisher's exact test for independence between the receptor sequence and the binary variable of interest.
- shared_by_n_samples
The number of samples in which the sequence was observed.
- samples_g0
Of the samples in which the sequence was observed, the number of samples belonging to the first group.
- samples_g1
Of the samples in which the sequence was observed, the number of samples belonging to the second group.
- shared_by_n_subjects
The number of subjects in which the sequence was observed (only present if subject IDs are specified).
- subjects_g0
Of the subjects in which the sequence was observed, the number of subjects belonging to the first group (only present if subject IDs are specified).
- subjects_g1
Of the subjects in which the sequence was observed, the number of subjects belonging to the second group (only present if subject IDs are specified).
- max_freq
The maximum clone frequency across all samples. Only present if
freq_colis non-null.- label
A character string summarizing the above information. Also includes the maximum in-sample clone frequency across all samples, if available.
References
Hai Yang, Jason Cham, Brian Neal, Zenghua Fan, Tao He and Li Zhang. (2023). NAIR: Network Analysis of Immune Repertoire. Frontiers in Immunology, vol. 14. doi: 10.3389/fimmu.2023.1181825
Searching for Associated TCR/BCR Clusters article on package website
Author
Brian Neal (Brian.Neal@ucsf.edu)
Examples
set.seed(42)
## Simulate 30 samples from two groups (treatment/control) ##
n_control <- n_treatment <- 15
n_samples <- n_control + n_treatment
sample_size <- 30 # (seqs per sample)
base_seqs <- # first five are associated with treatment
c("CASSGAYEQYF", "CSVDLGKGNNEQFF", "CASSIEGQLSTDTQYF",
"CASSEEGQLSTDTQYF", "CASSPEGQLSTDTQYF",
"RASSLAGNTEAFF", "CASSHRGTDTQYF", "CASDAGVFQPQHF")
# Relative generation probabilities by control/treatment group
pgen_c <- matrix(rep(c(rep(1, 5), rep(30, 3)), times = n_control),
nrow = n_control, byrow = TRUE)
pgen_t <- matrix(rep(c(1, 1, rep(1/3, 3), rep(2, 3)), times = n_treatment),
nrow = n_treatment, byrow = TRUE)
pgen <- rbind(pgen_c, pgen_t)
simulateToyData(
samples = n_samples,
sample_size = sample_size,
prefix_length = 1,
prefix_chars = c("", ""),
prefix_probs = cbind(rep(1, n_samples), rep(0, n_samples)),
affixes = base_seqs,
affix_probs = pgen,
num_edits = 0,
output_dir = tempdir(),
no_return = TRUE
)
#> [1] TRUE
## Step 1: Find Associated Sequences ##
sample_files <-
file.path(tempdir(),
paste0("Sample", 1:n_samples, ".rds")
)
group_labels <- c(rep("reference", n_control),
rep("comparison", n_treatment))
associated_seqs <-
findAssociatedSeqs(
file_list = sample_files,
input_type = "rds",
group_ids = group_labels,
seq_col = "CloneSeq",
min_seq_length = NULL,
drop_matches = NULL,
min_sample_membership = 0,
pval_cutoff = 0.1
)
head(associated_seqs[, 1:5])
#> ReceptorSeq fisher_pvalue shared_by_n_samples samples_g0 samples_g1
#> 8 CSVDLGKGNNEQFF 1.052106e-05 18 3 15
#> 7 CASSGAYEQYF 1.157316e-04 17 3 14
#> 4 CASSEEGQLSTDTQYF 5.197401e-03 10 1 9
#> 5 CASSIEGQLSTDTQYF 6.559548e-02 16 5 11
## Step 2: Find Associated Clones ##
dir_step2 <- tempfile()
findAssociatedClones(
file_list = sample_files,
input_type = "rds",
group_ids = group_labels,
seq_col = "CloneSeq",
assoc_seqs = associated_seqs$ReceptorSeq,
min_seq_length = NULL,
drop_matches = NULL,
output_dir = dir_step2
)
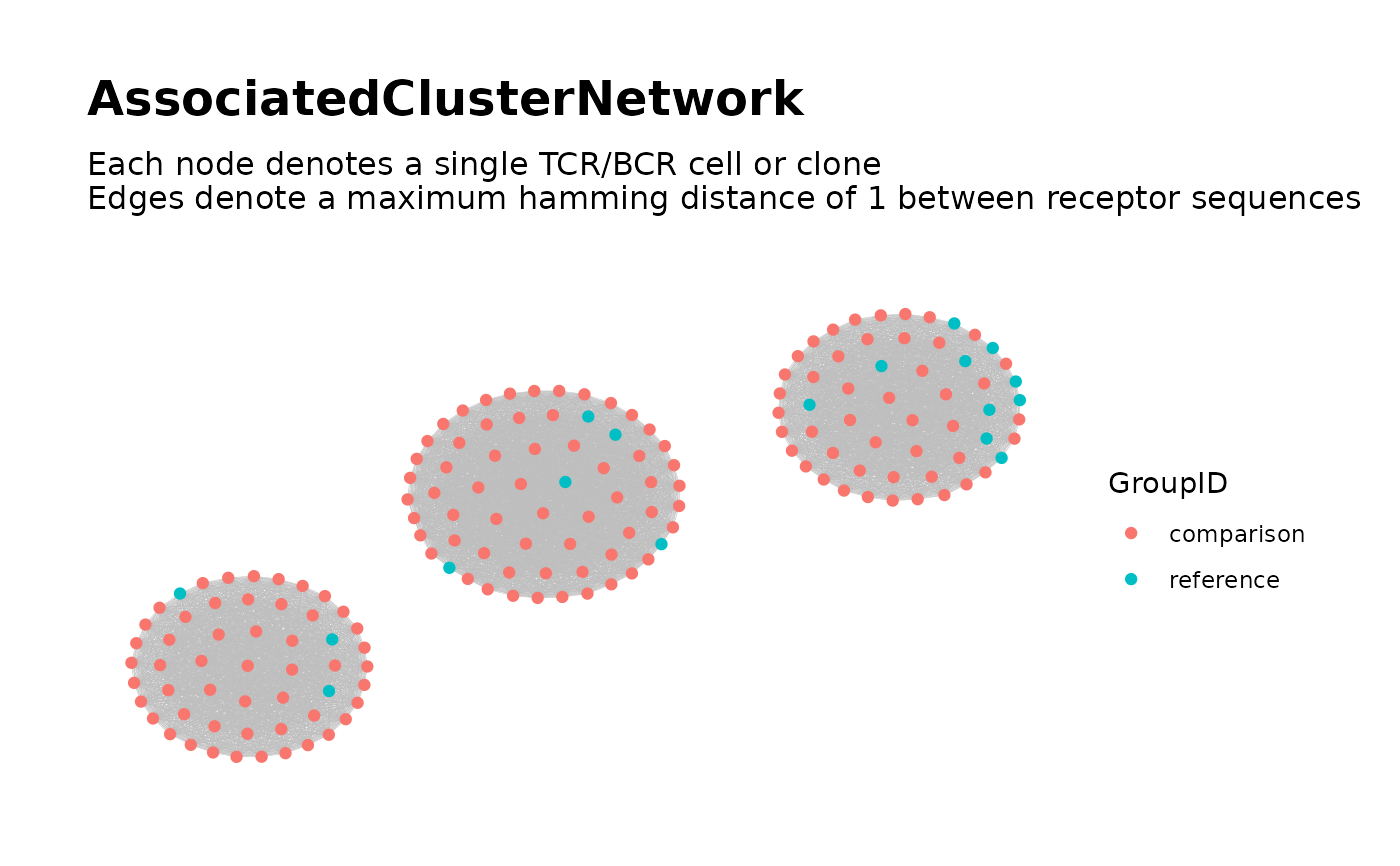
## Step 3: Global Network of Associated Clusters ##
associated_clusters <-
buildAssociatedClusterNetwork(
file_list = list.files(dir_step2,
full.names = TRUE
),
seq_col = "CloneSeq",
size_nodes_by = 1.5,
print_plots = TRUE
)